Second tutorial on GW¶
Treatment of metals.¶
This tutorial aims at showing how to obtain self-energy corrections to the DFT Kohn-Sham eigenvalues within the GW approximation, in the metallic case, without the use of a plasmon-pole model. The band width and Fermi energy of Aluminum will be computed.
The user may read the papers
-
F. Bruneval, N. Vast, and L. Reining, Phys. Rev. B 74 , 045102 (2006) [Bruneval2006], for some information and results about the GW treatment of Aluminum. He will also find there an analysis of the effect of self-consistency on quasiparticles in solids (not present in this tutorial, however available in Abinit). The description of the contour deformation technique that bypasses the use of a plasmon-pole model to calculate the frequency convolution of G and W can be found in
-
S. Lebegue, S. Arnaud, M. Alouani, P. Bloechl, Phys. Rev. B 67, 155208 (2003) [Lebegue2003],
with the relevant formulas.
A brief description of the equations implemented in the code can be found in the GW notes Also, it is suggested to acknowledge the efforts of developers of the GW part of ABINIT, by citing the 2005 ABINIT publication.
The user should be familiarized with the four basic tutorials of ABINIT, see the tutorial index as well as the first GW tutorial.
Visualisation tools are NOT covered in this tutorial. Powerful visualisation procedures have been developed in the Abipy context, relying on matplotlib. See the README of Abipy and the Abipy tutorials.
This tutorial should take about one hour to be completed (also including the reading of [Bruneval2006] and [Lebegue2003].
Note
Supposing you made your own install of ABINIT, the input files to run the examples are in the ~abinit/tests/ directory where ~abinit is the absolute path of the abinit top-level directory. If you have NOT made your own install, ask your system administrator where to find the package, especially the executable and test files.
To execute the tutorials, create a working directory (Work*) and
copy there the input files and the files file of the lesson. This will be explicitly mentioned in the first lessons,
that will tell you more about the files file (see also section 1.1).
The files file ending with _x (e.g. tbase1_x.files) must be edited every time you start to use a new input file.
Most of the tutorials do not rely on parallelism (except specific tutorials on parallelism). However you can run most of the tutorial examples in parallel, see the topic on parallelism.
In case you work on your own PC or workstation, to make things easier, we suggest you define some handy environment variables by executing the following lines in the terminal:
export ABI_HOME=Replace_with_the_absolute_path_to_the_abinit_top_level_dir export PATH=$ABI_HOME/src/98_main/:$PATH export ABI_TESTS=$ABI_HOME/tests/ export ABI_PSPDIR=$ABI_TESTS/Psps_for_tests/ # Pseudopotentials used in examples.
Examples in this tutorial use these shell variables: copy and paste the code snippets into the terminal (remember to set ABI_HOME first!). The ‘export PATH’ line adds the directory containing the executables to your PATH so that you can invoke the code by simply typing abinit in the terminal instead of providing the absolute path.
The preliminary Kohn-Sham band structure calculation¶
Before beginning, you might consider to work in a different subdirectory as for the other tutorials. Why not Work_gw2?
In tutorial 4, we have computed different properties of Aluminum within the DFT(LDA). Unlike for silicon, in this approximation, there is no outstanding problem in the computed band structure. Nevertheless, as you will see, the agreement of the band structure with experiment can be improved significantly if one relies on the GW approximation.
In the directory Work_gw2, copy the files tgw2_x.files and tgw2_1.in located in $ABI_TESTS/tutorial/Input, and modify the tgw2_x.files file as usual (see the first tutorial).
cd $ABI_TESTS/tutorial/Input mkdir Work_gw2 cd Work_gw2 cp ../tgw2_x.files . # modify this file as usual (see tutorial 1) cp ../tgw2_1.in .
Then issue:
abinit < tgw2_x.files > tgw2_1.log 2> err &
tgw2_x.in tgw2_x.out tgw2i tgw2o tgw2t ../../Psps_for_tests/13al.981214.fhi
# Crystalline aluminum # Create the WFK file for the GW calculation. ndtset 2 nband1 6 toldfe1 1.0d-8 iscf2 -2 getden2 1 nband2 35 nbdbuf2 5 tolwfr2 1.0d-10 #Definition of occupation numbers occopt 3 tsmear 0.05 #Definition of the unit cell acell 3*7.652 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 znucl 13 #Definition of the atoms natom 1 typat 1 xred 0.0 0.0 0.0 #Definition of the planewave basis set ecut 8.0 #Definition of the k-point grid ngkpt 4 4 4 #64 k points nshiftk 1 shiftk 0. 0. 0. istwfk *1 #256 k points #nshiftk 4 #shiftk 0 0 0 1/2 1/2 0 1/2 0 1/2 0 1/2 1/2 #istwfk 19*1 #Definition of the SCF procedure nstep 50 #toldfe 1.0d-8 prtvol 5 enunit 1 #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tgw2_1.in, tgw2_2.in, tgw2_3.in, tgw2_4.in #%% [files] #%% files_to_test = #%% tgw2_1.out, tolnlines=0, tolabs=0.000e+00, tolrel=3.000e-02 #%% psp_files = 13al.981214.fhi #%% [shell] #%% post_commands = #%% ww_cp tgw2_1o_DS2_WFK tgw2_2i_WFK; #%% ww_cp tgw2_1o_DS2_WFK tgw2_3i_WFK; #%% ww_cp tgw2_1o_DS2_WFK tgw2_4i_WFK; #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = F. Bruneval #%% keywords = GW #%% description = #%% Crystalline aluminum #%% Create the WFK file for the GW calculation. #%%<END TEST_INFO>
This run generates the WFK file for the subsequent GW computation and also provides the band width of Aluminum. Note that the simple Fermi-Dirac smearing functional is used (occopt = 3), with a large smearing (tsmear = 0.05 Ha). The k-point grid is quite rough, an unshifted 4x4x4 Monkhorst-Pack grid (64 k-points in the full Brillouin Zone, folding to 8 k-points in the Irreducible wedge, ngkpt = 4 4 4). Converged results would need a 4x4x4 grid with 4 shifts (256 k-points in the full Brillouin zone). This grid contains the \Gamma point, at which the valence band structure reaches its minimum.
The output file presents the Fermi energy
Fermi (or HOMO) energy (eV) = 7.14774 Average Vxc (eV)= -9.35982
as well as the lowest energy, at the \Gamma point
Eigenvalues ( eV ) for nkpt= 8 k points: kpt# 1, nband= 6, wtk= 0.01563, kpt= 0.0000 0.0000 0.0000 (reduced coord) -3.76175 19.92114 19.92114 19.92114 21.00078 21.00078
So, the occupied band width is 10.90 eV. More converged calculations would give 11.06 eV (see [Bruneval2006]). This is to be compared to the experimental value of 10.6 eV (see references in [Bruneval2006]).
Calculation of the screening file¶
In order not to lose time, let us start the calculation of the screening file before the examination of the corresponding input file. So, copy the file tgw2_2.in, and modify the tgw2_x.files file as usual (replace occurrences of twg2_x by tgw2_2). Also, copy the WFK file (tgw2_1o_WFK) to tgw2_2i_WFK. Then run the calculation (it should take about 30 seconds).
# Crystalline aluminum : # create the screening file #Parameter for the screening calculation optdriver 3 gwcalctyp 2 nband 30 ecuteps 4.0 ecutwfn 4.0 nfreqim 4 nfreqre 10 freqremax 1. #Definition of occupation numbers occopt 3 tsmear 0.05 #Definition of the unit cell acell 3*7.652 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 znucl 13 #Definition of the atoms natom 1 typat 1 xred 0.0 0.0 0.0 #Definition of the planewave basis set ecut 8.0 #Definition of the k-point grid ngkpt 4 4 4 nshiftk 1 shiftk 0. 0. 0. istwfk *1 #Definition of the SCF procedure nstep 50 toldfe 1.0d-8 prtvol 5 enunit 1 #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tgw2_1.in, tgw2_2.in, tgw2_3.in, tgw2_4.in #%% [files] #%% files_to_test = #%% tgw2_2.out, tolnlines= 32, tolabs= 1.1e-02, tolrel= 4.0e-01 #%% psp_files = 13al.981214.fhi #%% [shell] #%% post_commands = #%% ww_cp tgw2_2o_SCR tgw2_3i_SCR; #%% ww_cp tgw2_2o_SCR tgw2_4i_SCR #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = F. Bruneval #%% keywords = GW #%% description = #%% Crystalline aluminum: #%% create the screening file #%%<END TEST_INFO>
We now have to consider starting a GW calculation. However, unlike in the case of Silicon in the previous GW tutorial, where we were focussing on quantities close to the Fermi energy (spanning a range of a few eV), here we need to consider a much wider range of energy: the bottom of the valence band lies around -11 eV below the Fermi level. Unfortunately, this energy is of the same order of magnitude as the plasmon excitations. With a rough evaluation, the classical plasma frequency for a homogeneous electron gas with a density equal to the average valence density of Aluminum is 15.77 eV. Hence, using plasmon-pole models may be not really appropriate.
In what follows, one will compute the GW band structure without a plasmon-pole model, by performing explicitly the numerical frequency convolution. In practice, it is convenient to extend all the functions of frequency to the full complex plane. And then, making use of the residue theorem, the integration path can be deformed: one transforms an integral along the real axis into an integral along the imaginary axis plus residues enclosed in the new contour of integration. The method is extensively described in [Lebegue2003].
Examine the input file tgw2_2.in.
# Crystalline aluminum : # create the screening file #Parameter for the screening calculation optdriver 3 gwcalctyp 2 nband 30 ecuteps 4.0 ecutwfn 4.0 nfreqim 4 nfreqre 10 freqremax 1. #Definition of occupation numbers occopt 3 tsmear 0.05 #Definition of the unit cell acell 3*7.652 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 znucl 13 #Definition of the atoms natom 1 typat 1 xred 0.0 0.0 0.0 #Definition of the planewave basis set ecut 8.0 #Definition of the k-point grid ngkpt 4 4 4 nshiftk 1 shiftk 0. 0. 0. istwfk *1 #Definition of the SCF procedure nstep 50 toldfe 1.0d-8 prtvol 5 enunit 1 #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tgw2_1.in, tgw2_2.in, tgw2_3.in, tgw2_4.in #%% [files] #%% files_to_test = #%% tgw2_2.out, tolnlines= 32, tolabs= 1.1e-02, tolrel= 4.0e-01 #%% psp_files = 13al.981214.fhi #%% [shell] #%% post_commands = #%% ww_cp tgw2_2o_SCR tgw2_3i_SCR; #%% ww_cp tgw2_2o_SCR tgw2_4i_SCR #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = F. Bruneval #%% keywords = GW #%% description = #%% Crystalline aluminum: #%% create the screening file #%%<END TEST_INFO>
The ten first lines contain the important information. There, you find some input variables that you are already familiarized with, like optdriver, ecuteps, but also new input variables: gwcalctyp, nfreqim, nfreqre, and freqremax. The purpose of this run is simply to generate the screening matrices. Unlike for the plasmon-pole models, one needs to compute these at many different frequencies. This is the purpose of the new input variables. The main variable gwcalctyp is set to 2 in order to specify a non plasmon-pole model calculation. Note that the number of frequencies along the imaginary axis governed by nfreqim can be chosen quite small, since all functions are smooth in this direction. In contrast, the number of frequencies needed along the real axis set with the variable nfreqre is usually larger.
Finding the Fermi energy and the bottom of the valence band¶
In order not to lose time, let us start the calculation of the band width before the study of the input file. So, copy the file tgw2_3.in, and modify the tgw3_x.files file as usual (replace occurrences of twg2_x by tgw2_3). Also, copy the WFK file (tgw2_1o_WFK) to tgw2_3i_WFK, and the screening file (tgw2_2o_SCR) to tgw2_3i_SCR. Then run the calculation (it should take about 2 minutes on a 3 GHz PC).
The computation of the GW quasiparticle energy at the \Gamma point of Aluminum does not differ from the one of quasiparticle in Silicon. However, the determination of the Fermi energy raises a completely new problem: one should sample the Brillouin Zone, to get new energies (quasiparticle energies) and then determine the Fermi energy. This is actually the first step towards a self-consistency!
Examine the input file tgw2_3.in:
# Crystalline aluminum : # calculation of the quasi-particle Fermi energy #Parameters for the GW calculation optdriver 4 nband 30 ecutsigx 6.5 ecutwfn 6.5 gwcalctyp 12 symsigma 0 nkptgw 8 kptgw 0.000000 0.000000 0.000000 0.250000 0.000000 0.000000 0.500000 0.000000 0.000000 0.250000 0.250000 0.000000 0.500000 0.250000 0.000000 -0.250000 0.250000 0.000000 0.500000 0.500000 0.000000 -0.250000 0.500000 0.250000 bdgw 1 1 2 2 1 2 1 1 1 3 1 3 1 5 1 4 #Definition of occupation numbers occopt 3 tsmear 0.05 #Definition of the unit cell acell 3*7.652 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 znucl 13 #Definition of the atoms natom 1 typat 1 xred 0.0 0.0 0.0 #Definition of the planewave basis set ecut 8.0 #Definition of the k-point grid ngkpt 4 4 4 nshiftk 1 shiftk 0. 0. 0. istwfk *1 #Definition of the SCF procedure nstep 50 toldfe 1.0d-8 prtvol 5 enunit 1 #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tgw2_1.in, tgw2_2.in, tgw2_3.in, tgw2_4.in #%% [files] #%% files_to_test = #%% tgw2_3.out, tolnlines= 5, tolabs=0.002, tolrel= 3.000e-02 #%% psp_files = 13al.981214.fhi #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = F. Bruneval #%% keywords = GW #%% description = #%% Crystalline aluminum: #%% calculation of the quasi-particle Fermi energy #%%<END TEST_INFO>
The first thirty lines contain the important information. There, you find some input variables with values that you are already familiarized with, like optdriver, ecutsigx, ecutwfn. Then, comes the input variable gwcalctyp = 12. The value x2 corresponds to a contour integration. The value 1x corresponds to a self-consistent calculation with update of the energies only. Then, one finds the list of k points and bands for which a quasiparticle correction will be computed: nkptgw, kptgw, and bdgw. The number and list of k-points is simply the same as nkpt and kpt. One might have specified less k-points, though (only those needing an update). The list of band ranges bdgw has been generated on the basis of the DFT(LDA) eigenenergies. We considered only the bands in the vicinity of the Fermi level: bands much below or much above are likely to remain much or much above the Fermi region. In the present run, we are just interested in the states that may cross the Fermi level, when going from DFT to GW. Of course, it would have been easier to select an homogeneous range for the whole Brillouin zone, e.g. from 1 to 5, but this would have been more time-consuming.
In the output file, one finds the quasiparticle energy at \Gamma, for the lowest band:
--- !SelfEnergy_ee kpoint : [ 0.000, 0.000, 0.000, ] spin : 1 KS_gap : 0.000 QP_gap : 0.000 Delta_QP_KS: 0.000 data: !SigmaeeData | Band E_DFT <VxcDFT> E(N-1) <Hhartree> SigX SigC[E(N-1)] Z dSigC/dE Sig[E(N)] DeltaE E(N)_pert E(N)_diago 1 -3.762 -9.451 -3.762 5.689 -15.617 5.940 0.761 -0.313 -9.623 -0.173 -3.934 -3.988 ...
(the last column is the relevant quantity). The updated Fermi energy is also mentioned:
New Fermi energy : 2.558310E-01 Ha , 6.961515E+00 eV
The last information is not printed in case of gwcalctyp lower than 10.
Combining the quasiparticle energy at \Gamma and the Fermi energy, gives the band width, 10.404 eV. Using converged parameters, the band width will be 10.54 eV (see [Bruneval2006]). This is in excellent agreement with the experimental value of 10.6 eV.
Computing a GW spectral function, and the plasmon satellite of Aluminum¶
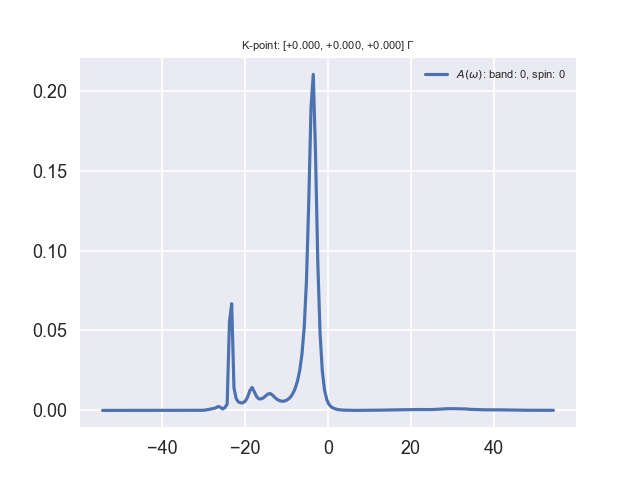
The access to the non-plasmon-pole-model self-energy (real and imaginary part) has additional benefit, e.g. an accurate spectral function can be computed, see [Lebegue2003]. You may be interested to see the plasmon satellite of Aluminum, which can be accounted for within the GW approximation.
Remember that the spectral function is proportional to (with some multiplicative matrix elements) the spectrum which is measured in photoemission spectroscopy (PES). In PES, a photon impinges the sample and extracts an electron from the material. The difference of energy between the incoming photon and the obtained electron gives the binding energy of the electron in the solid, or in other words the quasiparticle energy or the band structure. In simple metals, an additional process can take place easily: the impinging photon can extract an electron together with a global charge oscillation in the sample. The extracted electron will have a kinetic energy lower than in the direct process, because a part of the energy has gone to the plasmon. The electron will appear to have a larger binding energy… You will see that the spectral function of Aluminum consists of a main peak which corresponds to the quasiparticle excitation and some additional peaks which correspond to quasiparticle and plasmon excitations together.
In order not to lose time, this calculation can be started before the examination of the input file. So, copy the file tgw2_4.in, and modify the tgw4_x.files file as usual (replace occurrences of twg2_x by tgw2_4). Also, copy the WFK file (tgw2_1o_WFK) to tgw2_4i_WFK, and the screening file (tgw2_2o_SCR) to tgw2_4i_SCR. Then run the calculation (it should take about 2 minutes on a 3 GHz PC).
# Crystalline aluminum : perform the GW calculation # at the bottom of the valence band # Obtain the corresponding spectral function #Parameters for the GW calculation optdriver 4 gwcalctyp 2 symsigma 0 nband 30 ecutsigx 4.0 ecutwfn 4.0 nfreqsp 200 freqspmax 2. nkptgw 1 kptgw 0.000000 0.000000 0.000000 bdgw 1 1 icutcoul 3 # old deprecated value of icutcoul, only used for legacy #Definition of occupation numbers occopt 3 tsmear 0.05 #Definition of the unit cell acell 3*7.652 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 znucl 13 #Definition of the atoms natom 1 typat 1 xred 0.0 0.0 0.0 #Definition of the planewave basis set ecut 8.0 #Definition of the k-point grid ngkpt 4 4 4 nshiftk 1 shiftk 0. 0. 0. istwfk *1 prtvol 5 enunit 1 #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tgw2_1.in, tgw2_2.in, tgw2_3.in, tgw2_4.in #%% [files] #%% files_to_test = #%% tgw2_4.out, tolnlines= 2, tolabs= 1.010e-06, tolrel= 3.000e-02 #%% psp_files = 13al.981214.fhi #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = F. Bruneval #%% keywords = GW #%% description = #%% Crystalline aluminum : perform the GW calculation #%% at the bottom of the valence band #%% Obtain the corresponding spectral function #%%<END TEST_INFO>
Compared to the previous file (tgw2_3.in), the input file contains two additional keywords: nfreqsp, and freqspmax. Also, the computation of the GW self-energy is done only at the \Gamma point.
The spectral function is written in the file tgw2_4o_SIG. It is a simple text file. It contains, as a function of the frequency (eV), the real part of the self-energy, the imaginary part of the self-energy, and the spectral function. You can visualize it using your preferred software. For instance, start gnuplot and issue
p 'tgw2_4o_SIG' u 1:4 w l
You should be able to distinguish the main quasiparticle peak located at the GW energy (-3.7 eV) and some additional features in the vicinity of the GW eigenvalue minus a plasmon energy (-3.7 eV - 15.8 eV = -19.5 eV).
Tip
If AbiPy is installed on your machine, you can use the abiopen script
with the --expose option to visualize the results:
abiopen.py tgw2_4o_SIGRES.nc -e -sns
Another file, tgw2_4o_GW, is worth to mention: it contains information to be used for the subsequent calculation of excitonic effects within the Bethe-Salpeter Equation with ABINIT or with other codes from the ETSF software page.